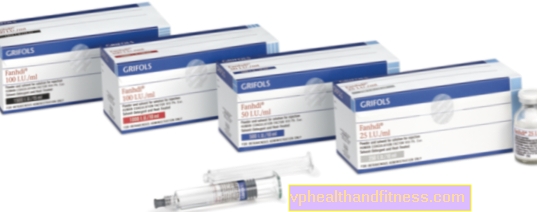
1 frasco contém 250 IU, 500 IU ou 1000 IU fator VIII de coagulação do sangue humano (FVIII) e 300 IU, 600 IU, respectivamente ou 1.200 UI o fator de von Willebrand humano. A atividade específica é de pelo menos 2,5 UI. até 10 UI FVIII: proteína C / mg dependendo do tamanho da embalagem (250 IU, 500 IU e 1000 IU). Produzido a partir do plasma de doadores humanos.
| Nome | Conteúdo da embalagem | A substância ativa | Preço 100% | Última modificação |
| FANHDI | 1 frasco + seringa de 1 ampere, pó e reconstituição para preparação solução para choque e / ou inf. | Fator VIII, Fator von Willebrand | 2019-04-05 |
Açao
Combinação de fatores de coagulação - fator VIII e fator de von Willebrand são componentes fisiológicos do plasma humano que atuam como componentes endógenos. A administração do fator de von Willebrand permite a correção de distúrbios hemostáticos que ocorrem em pacientes com deficiência do fator de von Willebrand em dois níveis: o fator de von Willebrand restabiliza a adesão das plaquetas ao endotélio vascular no local da lesão do vaso (ele se liga ao endotélio e à membrana das plaquetas sanguíneas), resultando no primário hemostasia, que se manifesta por uma redução no tempo de sangramento (o efeito aparece imediatamente e é amplamente dependente do nível de polimerização da proteína); O fator de von Willebrand causa um aumento retardado na deficiência concomitante de fator VIII, uma injeção intravenosa de fator de von Willebrand liga-se ao fator VIII endógeno (que é sintetizado no paciente), estabilizando este fator evita sua rápida degradação. Por este motivo, a administração de fator de von Willebrand puro (uma preparação de fator de von Willebrand de fator VIII baixo) restaura os níveis de fator VIII ao normal como efeito secundário após a primeira perfusão. Quando administrado por via intravenosa em pacientes com hemofilia, o fator VIII liga-se ao fator de von Willebrand na corrente sanguínea do paciente. O fator VIII ativo (VIlla) atua como cofator do fator IX (IXa) ativo para acelerar a conversão do fator X em fator X ativo (Xa). O fator X ativo converte a protrombina em trombina. A trombina então converte o fibrinogênio em fibrina, que permite a formação de um coágulo. Em pacientes com hemofilia A, após uma injeção, cerca de dois terços a três quartos do fator VIII permanecem na circulação. O nível de atividade do fator VIII atingido no plasma deve estar entre 80% e 120% da atividade do fator VIII prevista. A atividade do fator VIII no plasma diminui com uma distribuição exponencial em duas fases. Na fase inicial, a distribuição entre os compartimentos intravasculares e outros (fluidos corporais) ocorre com uma meia-vida de eliminação plasmática de 3 a 6 horas. h.
Dosagem
Por via intravenosa. O tratamento deve ser iniciado e supervisionado por um médico com experiência no tratamento de distúrbios hemorrágicos. Deficiência do fator VIII: a dose e a duração do tratamento dependem da gravidade da deficiência do fator VIIII, da localização e extensão do sangramento e da condição clínica do paciente. 1 UI A atividade do fator VIII é equivalente à quantidade de fator VIII em 1 ml de plasma humano normal. Administração de 1 UI fator VIII / kg aumenta a atividade do fator VIII plasmático em aproximadamente 1,7% - 2,5% da atividade normal, portanto, a dose necessária é calculada da seguinte forma: número necessário de unidades = peso corporal (kg) x aumento desejado do fator VIII (%) x 0,5 . A quantidade a ser administrada e a frequência de administração devem sempre depender da eficácia clínica individual em um determinado caso (às vezes, por exemplo, na presença de um baixo título de inibidor, pode ser necessário usar uma dose superior à calculada de acordo com a fórmula). No sangramento e na cirurgia, a atividade do fator VIII ao longo do tempo não deve cair abaixo dos seguintes níveis de atividade (em% do normal ou IU / dL): sangramento precoce nas articulações, músculos ou boca - o nível necessário do fator VIII é 20-40% do normal e o medicamento é administrado a cada 12-24 horas por pelo menos 1 dia, até que o sangramento passe (alívio da dor) ou a ferida esteja curada; sangramento mais grave nas articulações, músculos ou hematoma - o nível de fator VIII necessário é 30-60% do normal, e as infusões de medicamentos são repetidas a cada 12-24 horas por 3-4 dias ou mais até que o alívio da dor e o comprometimento agudo da função sejam resolvidos; sangramento com risco de vida - o nível de fator VIII necessário é 60-100% do normal, as infusões de medicamentos são repetidas a cada 8-24 horas até que a ameaça seja resolvida; pequena cirurgia (incluindo extração dentária) - o nível de fator VIII necessário é de 30-60%, o medicamento é administrado a cada 24 horas por pelo menos 1 dia, até que a ferida esteja curada; cirurgia de grande porte - os níveis de fator VIII necessários são de 80-100% (antes e após a cirurgia), as infusões são repetidas a cada 8-24 horas até que a ferida esteja curada, então continuados por pelo menos 7 dias consecutivos, mantendo os níveis de fator VIII em 30-60 % (IU / dL). A resposta ao tratamento varia individualmente e, portanto, o monitoramento dos níveis de fator VIII é essencial, especialmente em cirurgias de grande porte. Para a profilaxia de sangramento de longo prazo na hemofilia A grave, a dose usual de fator VIII é de 20-40 UI / kg. a cada 2-3 dias, intervalos de dose mais curtos ou doses mais altas podem ser necessários em pacientes mais jovens. Doença de Von Willebrand. É geralmente assumido que a administração de 1 UI de VWF: RCo / kg de peso corporal aumenta o nível de VWF: RCo na circulação em 2%. O objetivo do tratamento é obter o nível de VWF: RCo> 0,6 UI / ml (60%) e FVIII: C> 0,4 UI / ml (40%) no plasma. Na maioria dos casos, para a hemostasia, é recomendada uma dose de 40-80 UI / kg de peso corporal de fator de von Willebrand e 20-40 UI / kg de peso corporal de FVIII: C. Pacientes com doença de von Willebrand tipo 3 que podem requerer doses mais altas para manter níveis adequados do fator podem requerer uma dose inicial de fator de von Willebrand de 80 UI / kg de peso corporal. A dose selecionada deve ser administrada a cada 12-24 horas. A dosagem e a duração do tratamento dependem da condição clínica do paciente, da localização e da extensão do sangramento e do nível de FVW: RCo e FVIII: C. Durante o período de uso de preparações de fator VIII contendo fator de von Willebrand, o médico assistente deve levar em consideração a possibilidade de um aumento excessivo dos níveis de FVIII: C. Para evitar um aumento excessivo dos níveis de FVIII: C, deve ser considerada uma redução da dose e / ou uma extensão do intervalo entre as doses ou a utilização de medicamentos contendo VWF e uma quantidade inferior de fator VIII após 24-48 horas de tratamento. Apenas dados limitados de ensaios clínicos em crianças menores de 6 anos de idade estão disponíveis para a indicação acima - não use. Em crianças, nas indicações acima mencionadas, a titulação para eficácia clínica também é realizada calculando a dose de acordo com o peso corporal. O medicamento é administrado como injeção intravenosa lenta (a taxa de administração não deve exceder 10 ml / min).
Indicações
Para prevenir e controlar o sangramento em pacientes com hemofilia A (deficiência congênita do fator VIII de coagulação humana). Prevenção e controle de sangramento (incluindo sangramento cirúrgico) em pacientes com doença de von Willebrand (VWD) quando a terapia com desmopressina (DDAVP) é ineficaz ou contra-indicada. A preparação pode ser usada no tratamento da deficiência adquirida do fator VIII de coagulação humana.
Contra-indicações
Hipersensibilidade à substância ativa ou a qualquer um dos excipientes.
Precauções
São possíveis reações de hipersensibilidade do tipo alérgico. Os pacientes devem ser informados sobre os primeiros sinais de reações de hipersensibilidade, incluindo erupção cutânea, urticária extensa, aperto no peito, respiração ofegante, hipotensão até choque anafilático. Se ocorrerem tais sintomas, a administração do medicamento deve ser interrompida imediatamente e o tratamento apropriado instituído conforme apropriado. Os doentes tratados com fator VIII de coagulação humana devem ser cuidadosamente monitorizados quanto ao desenvolvimento de inibidores do fator VIII por observação clínica e avaliação de testes laboratoriais. O risco de desenvolver inibidores está correlacionado com a duração da exposição ao fator VIII, sendo o risco mais elevado durante os primeiros 20 dias de exposição. Em raras ocasiões, os inibidores também podem se desenvolver após os primeiros 100 dias de exposição. A formação recorrente (baixo título) de inibidores foi relatada em pacientes previamente tratados por pelo menos 100 dias com história de formação de inibidores após a mudança de um medicamento contendo fator VIII. Portanto, é recomendada a monitorização cuidadosa dos pacientes quanto à ocorrência de inibidores após qualquer mudança para o medicamento. Há um risco de complicações trombóticas com o uso de medicamentos com fator de von Willebrand, especialmente em pacientes com fatores de risco clínicos ou laboratoriais conhecidos. Por esse motivo, o monitoramento dos pacientes em risco é necessário para detectar os primeiros sinais de trombose. Além disso, as recomendações das diretrizes atuais para o tratamento e prevenção de tromboembolismo venoso devem ser seguidas. Durante o período de tratamento com um medicamento contendo fator VIII e fator de von Willebrand, o médico deve ter em consideração que o tratamento prolongado pode causar um aumento excessivo dos níveis de FVIII: C. Em doentes tratados com medicamentos contendo fator VIII e fator de von Willebrand, os níveis de FVIII: C devem ser monitorizados para evitar a persistência de níveis excessivos que podem aumentar o risco de complicações trombóticas. Pacientes com doença de von Willebrand, especialmente o tipo 3, podem desenvolver anticorpos neutralizantes (inibidores) ao fator de von Willebrand. Se os níveis de atividade plasmática esperados de VWF: RCo não forem atingidos ou o sangramento for controlado com doses apropriadas, um exame deve ser realizado para verificar a presença de um inibidor do fator de von Willebrand. Em pacientes com níveis elevados de inibidor, a terapia com fator de von Willbrand pode não ser eficaz e outras opções terapêuticas devem ser consideradas. Se um acesso venoso central for necessário, podem ocorrer infecções locais, bacteriemia e trombose do cateter. A vacinação apropriada (contra os vírus da hepatite A e B) deve ser considerada em pacientes que estão recebendo regularmente fator VIII de coagulação derivado do plasma.
Atividade indesejável
Hipersensibilidade ou reações alérgicas (angioedema, sensação de queimação e ardência no local da injeção, calafrios, rubor paroxístico, urticária generalizada, dor de cabeça, erupção cutânea, queda da pressão arterial, sonolência, náusea, inquietação, taquicardia, aperto no peito, alfinetes e agulhas, vômitos, respiração ruidosa), em alguns casos levando a anafilaxia grave com choque. Raramente foram observados aumentos de temperatura. Os pacientes com hemofilia A podem desenvolver anticorpos neutralizantes (inibidores) do fator VIII. Quando esses inibidores se desenvolvem, uma resposta clínica insuficiente ao tratamento é observada. Em casos muito raros, os pacientes com doença de von Willebrand, especialmente doença do tipo 3, podem desenvolver anticorpos neutralizantes (inibidores) do fator de von Willebrand. Se tais inibidores se desenvolverem, uma resposta clínica insuficiente ao tratamento é observada. Esses anticorpos podem estar intimamente associados a uma reação anafilática. Por esta razão, os pacientes que apresentam reações anafiláticas devem ser testados para inibidores. Nesses casos, é recomendado entrar em contato com um centro especializado para o tratamento de distúrbios hemostáticos. Existe um risco de complicações trombóticas quando o medicamento é utilizado em doentes com doença de von Willebrand com fatores de risco clínicos ou laboratoriais conhecidos.
Gravidez e lactação
A preparação deve ser usada durante a gravidez e amamentação apenas se claramente necessário (nenhuma experiência com o uso do medicamento devido à rara ocorrência de hemofilia A em mulheres).
Interações
Não há interações conhecidas da síndrome FVIII / VWF com outras preparações.
A preparação contém a substância: Fator VIII, Fator von Willebrand
Medicamento reembolsado: NÃO






---waciwoci-i-zastosowanie.jpg)