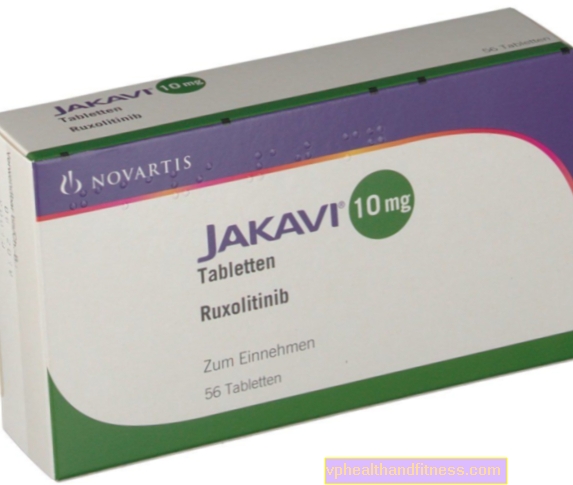
1 comprimido contém 5 mg, 10 mg, 15 mg ou 20 mg de ruxolitinib como fosfato. A preparação contém lactose.
| Nome | Conteúdo da embalagem | A substância ativa | Preço 100% | Última modificação |
| Jakavi | 56 pcs, mesa | Ruxolitinib | 2019-04-05 |
Açao
Um medicamento anticâncer, um inibidor da proteína quinase. O ruxolitinibe é um inibidor seletivo de Janus quinases (JAK), JAK1 e JAK2, que medeiam a sinalização para uma série de citocinas e fatores de crescimento que desempenham um papel importante na hemopoiese e na função imunológica. É caracterizado por alta permeabilidade, boa solubilidade e liberação rápida. É rapidamente absorvido após administração oral, a Cmax é atingida aproximadamente 1 hora após a administração. A ligação às proteínas plasmáticas é de cerca de 97%, principalmente à albumina. O ruxolitinib não atravessa a barreira hematoencefálica. É metabolizado principalmente pelo CYP3A4, com contribuição adicional do CYP2C9. No plasma, o fármaco está presente principalmente como fármaco inalterado e como dois metabólitos ativos. O ruxolitinib é eliminado principalmente pelo metabolismo. A eliminação média de T0,5 de ruxolitinib é de aproximadamente 3 horas e é excretada principalmente na urina e nas fezes.
Dosagem
Oralmente. O tratamento só deve ser iniciado por um médico com experiência na administração de medicamentos anticâncer. Um hemograma completo com um teste de glóbulos brancos deve ser realizado antes de iniciar o tratamento. O hemograma completo com esfregaço de leucócitos deve ser realizado a cada 2 a 4 semanas até que a dose esteja estabilizada e, então, dependendo das indicações clínicas. Dose inicial. Mielofibrose: 15 mg duas vezes ao dia em pacientes com contagem de plaquetas entre 100.000 / mm3 e 200.000 / mm3 e 20 mg duas vezes ao dia em pacientes com contagem de plaquetas> 200.000 / mm3. Policitemia Vera: 10 mg por via oral 2 vezes ao dia. A informação sobre a dose inicial recomendada para pacientes com contagem de plaquetas entre 50.000 / mm3 e 3 - máx. A dose inicial recomendada para esses pacientes é de 5 mg duas vezes ao dia e deve ser aumentada com cautela. Modificações de dose. As doses podem ser ajustadas com base na segurança e eficácia do medicamento. O tratamento deve ser interrompido se a contagem de plaquetas for inferior a 50.000 / mm3 ou a contagem absoluta de neutrófilos for inferior a 500 / mm3. O tratamento também deve ser interrompido em pacientes com PV se o nível de hemoglobina estiver abaixo de 8 g / dL. Após aumentar o número de hemogramas acima desses valores, a dosagem pode ser reiniciada com uma dose de 5 mg duas vezes ao dia com aumento gradual com base nos resultados de um teste de sangue completo com esfregaço. A redução da dose deve ser considerada se a contagem de plaquetas cair abaixo de 100.000 / mm3 para evitar a descontinuação do tratamento devido a trombocitopenia. A redução da dose também deve ser considerada em pacientes com PV se a hemoglobina estiver abaixo de 12 g / dL, e a redução da dose é recomendada se a hemoglobina estiver abaixo de 10 g / dL. Se o tratamento for considerado insuficientemente eficaz e o hemograma for adequado, a dose pode ser aumentada em um máximo de 5 mg duas vezes ao dia, até um máximo. doses de 25 mg duas vezes ao dia. A dose inicial não deve ser aumentada durante as primeiras quatro semanas de tratamento e, posteriormente, não deve ser aumentada com mais frequência do que em intervalos de 2 semanas. Máx. a dose da preparação é de 25 mg duas vezes ao dia. Ajustes de dose ao tomar concomitantes inibidores fortes do CYP3A4 ou fluconazol. A dose unitária deve ser reduzida em cerca de 50% e administrada duas vezes ao dia. Deve-se evitar o uso simultâneo da preparação com fluconazol em doses superiores a 200 mg ao dia. Durante o tratamento com inibidores fortes do CYP3A4 ou inibidores duplos das enzimas CYP2C9 e CYP3A4, recomenda-se o monitoramento mais frequente (por exemplo, duas vezes por semana) dos parâmetros hematológicos e dos sinais e sintomas de reações adversas relacionadas ao medicamento. Descontinuação do tratamento. O tratamento deve ser continuado enquanto a relação risco-benefício permanecer positiva, mas deve ser interrompido após 6 meses se não houver redução no tamanho do baço ou melhora dos sintomas desde o início do tratamento. Recomenda-se que os pacientes que mostram algum grau de melhora clínica interrompam o ruxolitinibe se apresentarem um alongamento do baço de 40% em comparação com o comprimento da linha de base (correspondendo aproximadamente a uma expansão do baço de 25%) e nenhuma melhora real do baço for observada. em relação aos sintomas associados à doença. Grupos especiais de pacientes. Nenhum ajuste especial da dose é necessário para pacientes com insuficiência renal leve ou moderada. Em pacientes com insuficiência renal grave (CCr 3 a 200.000 / mm3. Uma dose única de 20 mg ou 2 doses de 10 mg administradas com 12 horas de intervalo é recomendada para pacientes com MF com contagem de plaquetas> 200.000 / mm3. As doses subsequentes (administração única ou 2 doses de 10 mg administradas com 12 horas de intervalo) só devem ser administradas em dias de hemodiálise, após cada sessão de diálise. A dose inicial recomendada para pacientes em hemodiálise com ESRD e PV é uma dose única de 10 mg ou duas doses após cada sessão de diálise. 5 mg administrados em intervalos de 12 horas após a diálise e apenas no dia de hemodiálise Estas recomendações de dosagem são simuladas e quaisquer modificações de dose em pacientes com ESRD devem ser monitoradas cuidadosamente quanto à segurança e eficácia.Não existem dados disponíveis em pacientes em tratamento. diálise peritoneal ou hemofiltração veno-venosa contínua Em pacientes com qualquer insuficiência hepática, a dose inicial recomendada é baseada em e o as plaquetas devem ser reduzidas em aproximadamente 50% e administradas duas vezes ao dia. As doses subsequentes devem ser ajustadas com base na segurança e eficácia do medicamento. Os pacientes com diagnóstico de disfunção hepática durante o tratamento com a preparação devem fazer um exame de sangue completo com esfregaço de pelo menos 1 em 1-2 semanas durante as primeiras 6 semanas após o início do tratamento e, em seguida, após a estabilização da função hepática e exames de sangue - se disponíveis indicações clínicas. A dose pode ser ajustada para reduzir o risco de citopenia. Não são recomendados ajustes de dose adicionais em idosos. A segurança e eficácia em crianças com menos de 18 anos de idade não foram estabelecidas. Não há dados disponíveis. Forma de dar. Tome com ou sem comida. Se uma dose for esquecida, os pacientes não devem tomar uma dose extra, mas sim a próxima dose prescrita.
Indicações
Fibrose medular. Tratamento da esplenomegalia associada a uma doença ou sintomas observados em pacientes adultos com fibrose da medula óssea primária (também conhecida como fibrose da medula óssea idiopática crônica), mielofibrose precedida por policitemia (hiperemia) vera, ou fibrose da medula óssea precedida por trombocitemia essencial. Pato adornado com tufos. Tratamento de pacientes adultos com policitemia vera que são resistentes ou intolerantes à terapia com hidroxicarbamida.
Contra-indicações
Hipersensibilidade à substância ativa ou a qualquer um dos excipientes. Gravidez e lactação.
Precauções
O medicamento pode causar efeitos colaterais hematológicos, incluindo trombocitopenia, anemia e neutropenia, portanto, um teste de sangue completo com esfregaço de leucócitos é necessário antes de iniciar o tratamento. O tratamento deve ser descontinuado em pacientes com contagem de plaquetas inferior a 50.000 / mm3 ou contagem absoluta de neutrófilos inferior a 500 / mm3. Foi observado que os pacientes com baixa contagem de plaquetas (3) no momento do início da terapia têm maior probabilidade de desenvolver trombocitopenia durante o tratamento. A trombocitopenia é geralmente reversível e geralmente pode ser tratada com redução da dose ou suspensão temporária da preparação, mas uma transfusão de plaquetas pode ser necessária dependendo da indicação clínica. Pacientes que desenvolvem anemia podem precisar de transfusões de sangue, e ajustes de dose ou interrupção do tratamento podem ser considerados em pacientes com anemia. Pacientes com um nível de hemoglobina abaixo de 10,0 g / dL no início do tratamento têm um risco maior de níveis de hemoglobina abaixo de 8,0 g / dL durante o tratamento em comparação com pacientes com níveis basais de hemoglobina mais elevados, portanto, em pacientes com níveis basais de hemoglobina. hemoglobina abaixo de 10,0 g / dl, recomenda-se monitoramento mais frequente dos parâmetros hematológicos, bem como avaliação de sinais e sintomas indicativos de efeitos adversos associados ao uso da preparação. A neutropenia (contagem absoluta de neutrófilos <500 / mm3) foi geralmente reversível e controlável com a suspensão temporária do medicamento. Um teste de sangue completo deve ser realizado com a freqüência clinicamente indicada e ajuste de dose conforme necessário. Em pacientes tratados com a preparação ocorreram infecções bacterianas, micobacterianas, fúngicas, virais e outras infecções oportunistas graves, portanto, os pacientes devem ser avaliados quanto ao risco de infecções graves. Os médicos devem monitorar de perto os pacientes que recebem o medicamento quanto a sinais e sintomas de infecções e instituir o tratamento apropriado imediatamente. O tratamento não deve ser iniciado até que não haja mais uma infecção ativa grave. A tuberculose foi relatada em pacientes que tomam o medicamento para mielofibrose, e os pacientes devem ser testados para tuberculose ativa ou inativa (latente) antes de iniciar o tratamento, de acordo com as recomendações locais. As investigações devem incluir histórico médico, possíveis contatos anteriores com pacientes com tuberculose e / ou testes de triagem apropriados, como raio-x pulmonar, teste tuberculínico e / ou, se aplicável, teste de liberação de interferon-γ. Os prescritores devem ter em mente o risco de um teste tuberculínico falso-negativo, especialmente em pacientes gravemente enfermos ou imunocomprometidos. Aumentos no vírus da hepatite B (título de HBV-DNA), com ou sem aumentos concomitantes de ALT e AST, foram relatados em pacientes com infecção VHB crônica em uso do medicamento. O efeito da droga na replicação viral em pacientes com infecção VHB crônica é desconhecido; Os pacientes com VHB crônico devem ser tratados e monitorados de acordo com as diretrizes clínicas. Devido ao risco de herpes zoster, os médicos devem instruir os pacientes a reconhecer os sinais e sintomas precoces, recomendando que o tratamento seja iniciado o mais cedo possível. Leucoencefalopatia multifocal progressiva (PML) foi relatada com o uso de Jakavi para o tratamento de MF, portanto, os médicos devem estar vigilantes em relação aos sintomas que podem ser sugestivos de PML que os pacientes podem não notar (por exemplo, cognitivos, neurológicos ou mental). Os pacientes devem ser monitorados quanto a sintomas novos ou agravamento e, se tais sintomas se desenvolverem, um encaminhamento a um neurologista deve ser considerado ou devem ser instituídas contramedidas diagnósticas apropriadas. Se houver suspeita de PML, o tratamento adicional deve ser suspenso até que a PML seja excluída. Neoplasias não melanoma da pele (NMSC) (incluindo carcinoma basocelular, carcinoma de células escamosas e carcinoma de células de Merkel) foram relatadas em pacientes tratados com ruxolitinibe, a maioria desses pacientes com história de terapia de longo prazo com hidroxicarbamida e NMSC anterior ou lesões cutâneas pré-cancerosas. O exame periódico da pele é recomendado em pacientes com risco aumentado de câncer de pele. O tratamento com a preparação foi associado a aumentos nos parâmetros lipídicos, incluindo colesterol total, colesterol HDL, colesterol LDL e triglicerídeos - recomenda-se monitorar os níveis de lipídeos e tratar a dislipidemia de acordo com as diretrizes clínicas. A dose inicial deve ser reduzida em pacientes com insuficiência renal grave. Em pacientes com doença renal em estágio terminal e MF em hemodiálise, a dose inicial deve ser baseada na contagem de plaquetas; as doses subsequentes só devem ser administradas no dia de hemodiálise após o final de cada sessão de hemodiálise. Ajustes adicionais na dosagem devem ser feitos com monitoramento cuidadoso da segurança e eficácia do medicamento. A dose inicial deve ser reduzida em aproximadamente 50% em pacientes com insuficiência renal. Devem ser feitos ajustes posológicos adicionais com base na segurança e eficácia do medicamento. Se a preparação for administrada concomitantemente com inibidores fortes do CYP3A4 ou inibidores duplos das enzimas CYP3A4 e CYP2C9 (por exemplo, fluconazol), a dose unitária deve ser reduzida em cerca de 50% e administrada duas vezes ao dia. O uso concomitante de drogas citorredutoras ou fator de crescimento hematopoiético e a preparação não foi estudada. Após a interrupção ou descontinuação do tratamento, os sintomas de MF podem retornar em cerca de 1 semana. Existem casos conhecidos de pacientes que interromperam o tratamento com a preparação e apresentaram eventos mais graves, especialmente aqueles com outras comorbidades agudas. Não se sabe se a interrupção abrupta do tratamento contribuiu para a ocorrência desses eventos. A redução gradual da dose pode ser considerada, exceto quando a interrupção abrupta do tratamento for necessária, embora a utilidade da redução gradual da dose não tenha sido comprovada. A preparação contém lactose - não deve ser usada em pacientes com problemas hereditários raros de intolerância à galactose, deficiência de lactase de Lapp ou má absorção de glicose-galactose.
Atividade indesejável
Em pacientes com policitemia vera. Muito comuns: infecções do trato urinário, anemia de grau 3 (3) e grau 3 (50.000 - 25.000 / mm3) CTCAE, neutropenia de grau 3.(3) e 4 (3) CTCAE, hemorragia intracraniana, sangramento gastrointestinal, outro sangramento (incluindo epistaxe, hemorragias pós-operatórias e hematúria), flatulência, elevação de alanina aminotransferase de grau 3 CTCAE ( > 5x - 20x ULN). Incomum: tuberculose. Em pacientes com mielofibrose. Muito comuns: grau CTCAE qualquer anemia, grau CTCAE qualquer grau de trombocitopenia, sangramento (qualquer sangramento, incluindo sangramento intracraniano e gastrointestinal, hematomas e outros sangramentos, hematomas, outros sangramentos (incluindo epistaxe, pós-procedimento e hematúria), hipercolesterolemia CTCAE Grau 1 e 2, hipertrigliceridemia CTCAE Grau 1, tonturas, CTCAE Grau 1 alanina e elevação de aspartato de aminotransferase, hipertensão Comum: infecções do trato urinário, herpes zoster, trombocitopenia Grau 3 (50 CTCAE 000 - 25.000 / mm3), ganho de peso, constipação Incomum: elevação da anemia CTCAE Grau 3 (3) da alanina aminotransferase (> 5x - 20x LSN) Após a interrupção do tratamento em pacientes Com a MF, pode ocorrer recorrência dos sintomas da MF, como fadiga, dor óssea, febre, coceira, suores noturnos, aumento sintomático do baço e perda de peso. Em ensaios clínicos com MF, a pontuação total da pontuação dos sintomas de MF voltou gradualmente à linha de base no prazo de 7 dias após a interrupção do tratamento.
Gravidez e lactação
O uso da preparação durante a gravidez é contra-indicado. As mulheres com potencial para engravidar devem usar métodos contraceptivos eficazes durante o tratamento e, se uma mulher engravidar durante o tratamento com a preparação, deve ser feita uma avaliação individual de risco-benefício com aconselhamento sobre o possível risco para o feto. Não deve ser usado durante a amamentação e, portanto, a amamentação deve ser interrompida ao iniciar o tratamento.
Comentários
Tem efeito sedativo nulo ou insignificante. No entanto, os doentes que sentirem tonturas após tomarem a preparação devem evitar conduzir ou utilizar máquinas.
Interações
Os estudos de interação foram realizados apenas em adultos. O ruxolitinib é eliminado pelo metabolismo catalisado pelo CYP3A4 e CYP2C9, pelo que as preparações que inibem a atividade destas enzimas podem resultar no aumento da exposição ao ruxolitinib. Ao administrar o medicamento com inibidores fortes do CYP3A4 (tais como, mas não se limitando a, iboceprevir, claritromicina, indinavir, itraconazol, cetoconazol, lopinavir / ritonavir, ritonavir, mibefradil, nefazodona, nelfinavir, posaconavir, sa, sa, 50% e administrado duas vezes ao dia. Os pacientes devem ser monitorados de perto (por exemplo, duas vezes por semana) para possível citopenia, e a dose deve ser aumentada gradualmente com base na segurança e eficácia. Deve ser considerada uma redução da dose de 50% ao usar preparações que são inibidores duplos das enzimas CYP2C9 e CYP3A4 (por exemplo, fluconazol) .O uso concomitante do fármaco com fluconazol em doses superiores a 200 mg por dia deve ser evitado. Ao tomar indutores do CYP3A4 (tais como, mas não se limitando a avasimibe, carbamazepina, fenobarbital, fenitoína, rifabutina, rifampicina (rifampicina), erva de São João (Hypericum perforatum), os pacientes devem ser monitorados de perto e a dose deve ser aumentada gradualmente com base na segurança e eficácia. Nenhum ajuste de dose é recomendado quando ruxolitinib é administrado concomitantemente com inibidores leves ou moderados do CYP3A4 (tais como, mas não se limitando a, ciprofloxacina, eritromicina, amprenavir, atazanavir, diltiazem, cimetidina), no entanto, os pacientes devem ser monitorados de perto para possível citopenia ao iniciar a terapia com inibidores moderados. CYP3A4: O ruxolitinib pode inibir a glicoproteína P intestinal e a proteína de resistência ao cancro da mama (BCRP), o que pode resultar no aumento da exposição sistémica de substratos destes transportadores, como etexilato de dabigatrano, ciclosporina, rosuvastatina e potencialmente digoxina. monitoramento de drogas (TDM) ou condição clínica após a administração das substâncias mencionadas. É possível que a inibição potencial de P-gp e BCRP no intestino seja minimizada se o tempo entre as administrações do medicamento for mantido o maior possível. O uso concomitante de fatores de crescimento hematopoiéticos e a preparação não foi estudado. Não se sabe se a inibição de Janus quinases (JAK) por Jakavi reduz a eficácia dos fatores de crescimento hematopoiéticos ou se os fatores de crescimento hematopoiéticos afetam a eficácia da preparação. O uso simultâneo de terapias citorredutoras e da preparação não foi estudado - a segurança e a eficácia da administração simultânea desses medicamentos são desconhecidas. O ruxolitinib não inibe o metabolismo do midazolam, substrato do CYP3A4 oral - portanto, não é esperado nenhum aumento na exposição aos substratos do CYP3A4 quando estes medicamentos são coadministrados com Jakavi. A preparação não afeta a farmacocinética de um contraceptivo oral contendo etinilestradiol e levonorgestrel, portanto, não se espera que a eficácia de contraceptivos contendo esta combinação seja reduzida durante o uso concomitante de ruxolitinib.
A preparação contém a substância: Ruxolitinib
Medicamento reembolsado: NÃO