As encefalopatias espongiformes (doenças de príons) são aquelas em que formas patológicas de proteínas de príons estão envolvidas no desenvolvimento. Sabemos cada vez mais sobre as doenças por príons, mas os principais aspectos permanecem desconhecidos - no momento, a medicina não tem os meios para curar os pacientes dessas doenças.
As encefalopatias espongiformes, ou doenças priônicas, podem se desenvolver durante a vida, enquanto outras surgem de mutações genéticas hereditárias presentes desde o nascimento. Dentro deste grupo, existem várias entidades que ocorrem em humanos, como exemplos a doença de Creutzfeldt-Jakob ou a insônia familiar fatal.
As doenças por príons são muito misteriosas há muito tempo. Ao contrário de outros patógenos, como bactérias, vírus ou fungos, eles não contêm ácido nucléico - os príons são feitos apenas de proteínas. A teoria das doenças por príons foi descoberta por S. Prusiner, essa descoberta foi muito apreciada pela comunidade científica - em 1997 o pesquisador recebeu o Prêmio Nobel de Medicina. Embora relativamente muitos anos tenham se passado desde o nascimento do conceito de príon, alguns cientistas ainda acreditam que ele está incompleto e estão investigando mais a natureza dessas condições - alguns dos fatores responsáveis pelas encefalopatias espongiformes já foram confirmados.
Doenças de príon: causas
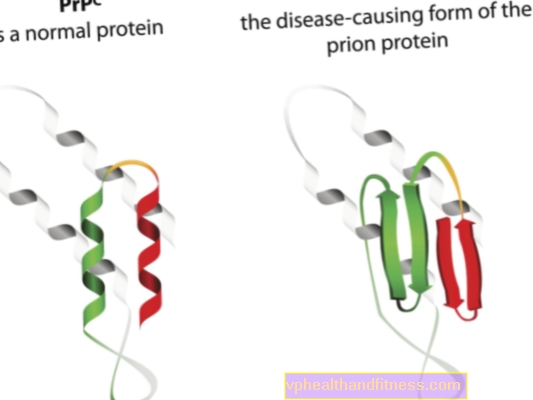
A etiologia das doenças por príons está relacionada à transformação de proteínas príon normais em formas patogênicas e patogênicas. Os príons são moléculas de proteínas encontradas no corpo de cada ser humano. Sua função ainda não está totalmente esclarecida, mas sabe-se que, em condições normais, as proteínas príon não prejudicam o corpo. No entanto, quando os príons mudam sua estrutura e se tornam partículas patogênicas, uma das várias encefalopatias espongiformes se desenvolve. Os príons que ocorrem naturalmente no corpo são chamados de PRPC, enquanto as formas anormais são chamadas de PRPSC. Estes últimos são um problema sério não só porque podem se acumular no tecido nervoso na forma de depósitos e gerar seus danos, mas também porque têm a capacidade de transformar príons normais em uma forma malformada (simplificando, PRPSC pode "infectar" proteínas normais com seu potencial patogênico).
Leia também: Doença de Huntington (coreia de Huntington): causas, sintomas, tratamento Tremores musculares - causas. O que significa tremor muscular? Doenças que matam mais rápido: CHOQUE, EBOLA, DAMN, ATAQUE, EMERGÊNCIA [GALE ...Basicamente, existem 3 causas de encefalopatia espongiforme:
- esporádico (mutação patogênica ocorre em células somáticas, ocorre durante a vida do paciente),
- família (resultante da carga de mutações herdadas dos pais),
- Passagem (relacionada à introdução de príons patogênicos no corpo humano, por exemplo, através de preparações de hormônio do crescimento contaminado com essas partículas ou transplante de córnea de uma pessoa que sofre de alguma encefalopatia espongiforme).
Encefalopatias espongiformes: doença de Creutzfeldt-Jakob
A doença de Creutzfeldt-Jakob (CJD) foi descrita pela primeira vez no início dos anos 1920. Existem 4 tipos de doença:
- CJD esporádica (o mais comum, responsável por até 9/10 de todos os casos de CJD)
- cidade natal de CJD
- CJD com cinto
- variante de CJD
O quadro clínico no curso de várias variantes da doença de Creutzfeldt-Jakob pode ser variável. As doenças mais comuns no curso deste grupo de encefalopatias espongiformes são:
- transtornos demenciais (incluindo deterioração progressiva da memória, atenção e concentração)
- mioclonia (movimentos involuntários, como movimentos bruscos dos músculos)
- disfunção cerebelar (manifestada, por exemplo, por distúrbios do equilíbrio)
- visão embaçada
- sintomas piramidais e extrapiramidais
No decurso das variantes da CJD, também podem aparecer distúrbios mentais (por exemplo, ansiedade, humor deprimido), dor e outros movimentos involuntários diferentes dos mencionados acima.
O prognóstico para a doença de Creutzfeldt-Jakob é ruim - por exemplo, em pacientes com DCJ esporádica, leva em média de quatro a cinco meses desde o início dos sintomas da doença até a morte.
Encefalopatias espongiformes: síndrome de Gerstmann-Straussler-Scheinker
A síndrome de Gerstmann-Straussler-Scheinker (GSS) geralmente ocorre em famílias e é causada por uma mutação hereditária no gene PRNP. É considerada a encefalopatia espongiforme de evolução mais lenta. A equipe GSS inclui:
- ataxia espinocerebelar
- disartria
- transtornos demenciais
- distúrbios de deglutição
- nistagmo
- aumento da tensão muscular
Os pacientes com diagnóstico de GSS têm um período de tempo variável e, em alguns pacientes, a morte ocorre mais de 10 anos após o início.
Encefalopatias espongiformes: insônia familiar fatal
A insônia familiar letal é uma doença priônica causada por mutações no gene PRNP. A doença é extremamente rara e até agora foi diagnosticada em 28 famílias em todo o mundo. No curso de uma insônia familiar fatal, o primeiro sintoma é a incapacidade de dormir. Esse problema resulta em transtornos de ansiedade e no paciente tendo alucinações. O efeito da constante falta de descanso noturno é a disfunção do sistema autônomo (incluindo alterações na função cardíaca, sudorese e distúrbios do sistema digestivo), há também uma diminuição progressiva do peso corporal. Em estágios mais avançados de insônia familiar fatal, aparecem distúrbios hormonais e ocorrem sintomas de demência durante o curso da doença.
O prognóstico para insônia familiar fatal, como para outras encefalopatias espongiformes, é ruim: os pacientes geralmente morrem dentro de três anos do início.
Encefalopatias espongiformes: prionopatia com susceptibilidade variável à protease
A ocorrência dessas encefalopatias espongiformes está relacionada principalmente a mutações no gene PRNP. No entanto, essas mutações dizem respeito a diferentes códons desse gene e, portanto, várias doenças de príons diferentes são distinguidas. Uma unidade descrita recentemente (em 2008) é a prionopatia com suscetibilidade variável à protease. Pessoas que sofrem desta doença carregam mutações em até três códons do gene PRNP.
Na prionopatia com susceptibilidade variável à protease, os pacientes experimentam:
- deficiência cognitiva
- extrema gravidade dos transtornos psiquiátricos: eles podem ser euforia e agitação, mas também apatia significativa
- disartria
- afasia (distúrbios das funções da linguagem)
A duração média da doença nesta prionopatia é inferior a 4 anos.
Encefalopatias espongiformes: kuru
O kuru é hoje considerado uma doença que praticamente não existe mais - foi encontrado em representantes de tribos de Papua-Nova Guiné, que praticavam comportamento canibal. O sintoma dominante dessa encefalopatia espongiforme é a ataxia cerebelar progressiva. Pode ser acompanhada por movimentos involuntários (principalmente na forma de coreia, tremores e atetose), bem como incontinência urinária e fecal. Pacientes em kuru também experimentam alterações de humor significativas, eles desenvolvem reflexos primitivos (por exemplo, sucção). Um problema bastante característico no caso dessa doença de príon são ataques forçados de choro ou riso - devido ao último fenômeno, o kuru às vezes é chamado de "morte sorridente".
Encefalopatias espongiformes: diagnóstico
As doenças por príons podem ser suspeitadas com base nos sintomas do paciente. No entanto, são bastante inespecíficos, pois também podem aparecer no curso de uma série de outras doenças não relacionadas aos príons. Por este motivo, os seguintes também são usados no diagnóstico de encefalopatias espongiformes:
- testes de imagem (por exemplo, imagem de ressonância magnética, que permite detectar alterações relacionadas à degeneração do cérebro por proteínas príon),
- testes de laboratório (como a avaliação das concentrações de proteínas no líquido cefalorraquidiano, por exemplo, MAP-tau, S-100 ou proteínas 14-3-3),
- testes genéticos (para detectar a presença de mutações no paciente),
- testes imunohistoquímicos (usando anticorpos para proteínas príon).
O diagnóstico também pode ser confirmado por autópsia do cérebro, na qual é possível encontrar alterações características das encefalopatias espongiformes. Estas podem ser lesões esponjosas, de distribuição variada e com uma estrutura diferente (dependendo da entidade específica da doença), placas amilóides e defeitos neuronais.
Encefalopatias espongiformes: tratamento
As doenças por príons são atualmente incuráveis - apesar dos numerosos estudos que vêm acontecendo há muitos anos, a medicina ainda não possui drogas que possam retardar ou inibir completamente seu progresso. O tratamento sintomático é utilizado em pacientes com encefalopatias espongiformes, com o objetivo de aliviar a intensidade dos sintomas e melhorar ao máximo sua qualidade de vida.
No entanto, o trabalho sobre o tratamento das encefalopatias espongiformes ainda está em andamento. Os cientistas estão tentando usar vários métodos - o primeiro exemplo é a terapia genética. Eles afetariam os ácidos nucléicos e as mutações presentes em sua estrutura - o objetivo da aplicação da terapia gênica seria neutralizar erros no código genético. Uma abordagem diferente é a base da terapia imunológica - o trabalho está em andamento para criar anticorpos cujo papel seria eliminar os príons patogênicos. Outro método que vê potencial para combater as encefalopatias espongiformes é o tratamento com o uso de moléculas de proteínas sintetizadas que, uma vez introduzidas no corpo do paciente, neutralizariam as proteínas patológicas.
Artigo recomendado:
Encefalopatias - causas, tipos e sintomas






-przyczyny-rozpoznanie-i-leczenie-napiciowego-blu-gowy.jpg)